

化学合成多肽药物药学研究技术
指导原则(试行)
2023年2月
一、前言
二、原料药制备工艺
(一)合成工艺的选择
(二)物料及控制
(三)生产工艺
1、工艺描述
2、反应终点控制
3、中间体控制
三、结构确证
四、制剂处方工艺
(一)剂型的选择
(二)处方筛选及工艺开发
(三)工艺描述及中间体控制
五、质量研究与控制
(一)原料药的质量研究
1、鉴别
2、氨基酸比值
3、杂质
4、含量测定
(二)制剂的质量研究
六、稳定性研究
七、参考文献
一、前言
多肽是由多个氨基酸通过酰胺键相连形成的化合物,可 通过基因重组表达、生物提取、化学合成等方法制备。
化学合成多肽药物在制备方法、结构确证、质量研究等 方面均存在自身特性,不能简单的将其归属于小分子化学药 物或生物制品。同时,许多国内外指导性文件均明确不涵盖 多肽类药物。本指导原则在2007年版《合成多肽药物药学研 究技术指导原则》的基础上,对合成多肽药物药学研究方面 所涉及的特殊问题进行分析,结合国内外对多肽药物研究和 评价的实践经验,提出化学合成多肽药物药学研究的一般性 技术要求,供业界参考执行。
本指导原则的起草是基于对多肽药物的当前认知,随着 多肽药物研究技术的提高和相关法规的完善,本指导原则将 不断修订。本指导原则仅适用于采用化学合成法制备的多肽药物。
二、原料药制备工艺
(一)合成工艺的选择
根据反应载体不同,多肽原料药的合成工艺主要包括液 相合成法和固相合成法。根据肽键形成的顺序,多肽原料药 的合成工艺主要包括线性合成法和片段缩合合成法。在合成 2 / 15 工艺选择时应结合目标肽链的长短和氨基酸特性综合考虑。 需要注意的是,在多肽合成过程中会产生一些与目标肽链结 构类似的杂质,如缺失肽、插入肽、错结肽和差向肽等,诸如此类杂质的产生和蓄积会增加多肽成品提纯的难度,合成 工艺选择时应避免或减少此类肽相关杂质的产生。
以固相合成工艺为例,在工艺开发时应关注氨基酸保护/ 脱保护策略、缩合剂选择依据、树脂选择依据、氨基酸偶联、 裂解、纯化、肽链修饰和富集冻干等研究内容。对于多肽纯 化方法的选择,需要综合考虑肽链长短、极性和荷电性以及 粗品纯度等因素,对纯化前后样品进行质量对比研究,证明 拟采用的纯化方式可有效起到提纯效果。在开发冻干工艺时 需充分考虑冻干条件的合理性,提供详细的冻干工艺参数, 并对冻干前后样品进行质量对比研究。过滤等工艺中若存在 极端pH值或有机溶剂条件,还需考虑进行必要的滤膜相容性 研究。
(二)物料及控制
根据 ICH Q11 及其问答文件等相关要求,在多肽原料药 的合成工艺中,保护氨基酸或肽段的质量状况直接影响多肽 药物质量,应定义为起始物料。
保护氨基酸的质量控制项目一般应包括性状、有关物质、 手性异构体、残留溶剂、含量测定等。由于保护氨基酸中的 杂质可能伴随化学反应而发生杂质传递和转化,继而增加多 3 / 15 肽药物的提纯难度,应增加必要的杂质研究,同时制定合理 的控制限度。应结合保护氨基酸的合成工艺确定杂质谱组成, 以 Fmoc 保护氨基酸为例,常见的杂质包括保护不完全氨基 酸、手性异构体、同分异构体、二肽衍生物、β-丙氨酸杂质 等(如表 1)。应开发出能有效检出各潜在杂质的分析方法, 并对分析方法进行必要的方法学验证,充分证明所采用方法 的可行性。保护氨基酸合成过程中如果使用了某些溶剂,如 乙酸乙酯、乙酸等,需要对其残留进行合理控制,以避免在 后续偶联反应中出现“封端”现象。
表 1 Fmoc 保护氨基酸中常见的杂质

片段缩合合成法中,采用肽段作为起始物料,应提供肽 段的合成工艺及必要的结构确证资料,应结合多肽终产品质 量标准制定肽段的质控项目,如鉴别、纯度和含量测定等。 肽段有关物质考察应重点关注可能引入到终产品中的其他 短肽残留情况,建立相应的杂质控制策略,并提供必要的方 法学验证资料。同时,也应该提供研究资料明确肽段的储存 条件和时限。
固相合成中,树脂作为必需物料,其质量可直接影响多 肽药物质量,应提供树脂的来源、质量标准和检验报告,并 对树脂的摩尔取代系数、所用合成条件下的膨胀系数和稳定 性情况等予以说明。由于缺乏有效的表征手段和针对性的质 控方法,不推荐与肽链C端氨基酸相连的树脂作为起始物料。
(三)生产工艺
1、工艺描述
应提供详细的工艺描述,以代表性批次,如注册批、工 艺验证批为例,按照工艺流程详细描述各步工艺操作,包括 反应试剂、投料量、反应条件、过程控制等。如果纯化方法 使用制备型液相色谱仪,应提供载样量、洗脱程序、收集范 围以及各洗脱液的处理方式等。
2、反应终点控制
液相合成反应终点一般可采用色谱法(如 TLC 法、HPLC 法)监测;固相合成反应的终点可采用茚三酮法、三硝基苯 磺酸法和四氯苯醌法等监测;二硫键连接的反应终点可采用 Ellman 试验或 HPLC 法监测。除上述方法外,其他合适的方 法也可以使用。监测方法应确保方法灵敏度符合检测要求。 需要注意的是,有些情况下单独一种监测方法的检测灵敏度 可能不够,可采用多种手段进行监测。
3、中间体控制
对于液相合成,应参照《化学药物原料药制备和结构确 5 / 15 证研究的技术指导原则》对中间体的质量进行控制,特别要 对缩合片段制订合理的质量控制措施(例如分子量、有关物 质和含量等)。
对于固相合成,在氨基酸偶联过程中,因肽链未与树脂 裂解,缺少有效的中控手段,对肽链从树脂切割后的中间体 应建立合理的控制措施。在综合分析后续纯化和冻干等步骤 杂质谱变化情况的基础上,制定合理的中间体质控限度.
三、结构确证
多肽分子由多种氨基酸组成,通常分子量较大,通过小 分子化学药物结构研究常用的一些方法(例如紫外光谱、红 外光谱、核磁共振波谱等)来解析多肽的结构存在一定的困 难。可采用适当的方法确证多肽药物一级结构(即氨基酸序 列及其修饰情况),并酌情采用其他方法确证其高级结构。
多肽原料药的一级结构确证中,应明确各氨基酸的连接 顺序。对于具有氨基酸侧链修饰的多肽以及存在两个以上分 子内环(如二硫键、酰胺键等)的环肽,应明确具体修饰位 点、修饰物结构以及每个内环键的具体连接位点。氨基酸序 列分析可采用多级质谱联用(MS-MS)、Edman降解等方法。
多肽药物的高级结构一般包括二级结构(如螺旋、折叠、 转角等)和三级结构,且与肽链所处的溶液环境密切相关。 可先通过文献资料等论述说明是否存在高级结构,如存在, 应结合药物制剂进行综合考察。对于具有一定高级结构才能发挥其生物活性或缓释作用的多肽创新药物,应分析其高级 结构对体内生物活性的影响。对于多肽仿制制剂,应具有与 参比制剂一致的高级结构。具体的研究方法例如:圆二色谱 (CD)、傅里叶变换红外光谱(FTIR)、拉曼光谱、核磁共 振波谱(NMR)、晶体X-射线衍射(XRD)、分析超速离心 (AUC)、场流分离(FFF)、荧光光谱等。同时,可结合多 肽药物的体内/体外生物活性研究结果,佐证其高级结构。
四、制剂处方工艺
化学合成多肽药物制剂研究应遵循小分子化学药物的 一般规律,可参照《化学药物制剂研究基本技术指导原则》、 《化学药品注射剂仿制药质量和疗效一致性评价技术要求》 等开展相应研究。同时,需关注多肽药物的特殊性,如理化 性质和稳定性等。
(一)剂型的选择
与小分子化学药物相比,多肽药物多具有稳定性差、易 酶解、脂溶性差等特点,化学合成多肽药物通常选择注射途 径给药。近年来,随着多肽药物研发的深入和制剂技术的进 步,多肽药物剂型选择的范围逐步扩大,鼓励根据临床使用 需要,开发临床需求广泛、顺应性好的剂型,如长效制剂、 口服制剂、吸入制剂等。
(二)处方筛选及工艺开发
在进行多肽药物的制剂研究时,需要充分了解原料药的 理化性质,如溶解度、pH 值、pI、比旋度、高级结构、水分、 油/水分配系数等,应关注原料药在不同 pH 值、离子强度、 光、热、湿、氧化等条件下的稳定性情况。结合拟开发药物 的关键质量属性,有针对性地进行研究。在整个处方工艺开 发过程中需关注多肽药物的化学和物理稳定性。
处方筛选时,可根据原料药性质和剂型特点进行辅料选 择,应有利于提高多肽药物的稳定性,避免辅料与原料药的 不良相互作用,同时应尽量减少不必要辅料的使用。关注辅 料是否会引起多肽药物的杂质谱和/或高级结构发生变化,从 而影响药物的安全性。例如,某些辅料中的醛基可能与肽链 中的氨基发生反应;某些辅料可能引起体系的 pH 值、离子 强度等发生变化,从而导致降解杂质增加、高级结构变化等。
工艺开发时,应充分了解引起多肽药物不稳定的因素, 在拟定生产工艺中应尽量避免多肽药物长期或短期处于不 稳定条件下。表 2 列出了制剂工艺开发中部分需要考虑的不 稳定因素。例如,配液过程中使用 pH 调节剂,可能会导致 局部 pH 值过高/过低,进而引起多肽药物降解等问题;对于 主药含量较低的多肽药物制剂,应关注容器、滤膜等的吸附 情况;粉针剂的冻干过程可能存在缓冲剂结晶、pH 值移位、 稳定剂形态改变、高级结构改变等问题;多肽药物多具有引湿性,应关注辅料中的水分、生产过程的湿度控制等;注射 使用的多肽药物,应关注无菌保障水平等。
表 2 多肽药物的不稳定因素
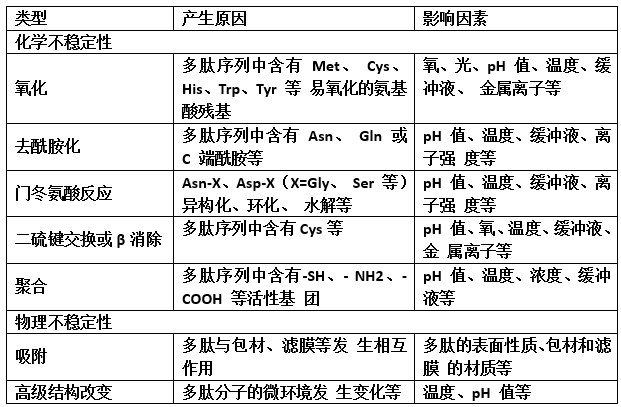
(三)工艺描述及中间体控制
工艺描述中应结合工艺开发时确定的关键生产步骤,提 供工艺操作细节,以保证制剂的批内和批间质量一致性。 结合药物生产过程,制定必要的中间体控制措施,建立 合理的控制限度,以保证生产可持续进行。多肽药物多为注 射剂,需对生物负载进行常规中控监测,对中间体存放条件 和时限进行研究。
五、质量研究与控制
(一)原料药的质量研究
合成多肽原料药的质量研究除参考小分子化学药物的 研究思路进行常规项目的研究外,还应根据合成多肽的制备 工艺、结构和理化特性等增加一些特定的考察项目,例如氨 基酸组成分析、反离子含量等。表 3 列举了合成多肽原料药 的质控项目作为参考,仅作为示例展示,并不代表全部情况, 应根据产品特性拟定具体考察项目。
表 3 合成多肽原料药的质控项目
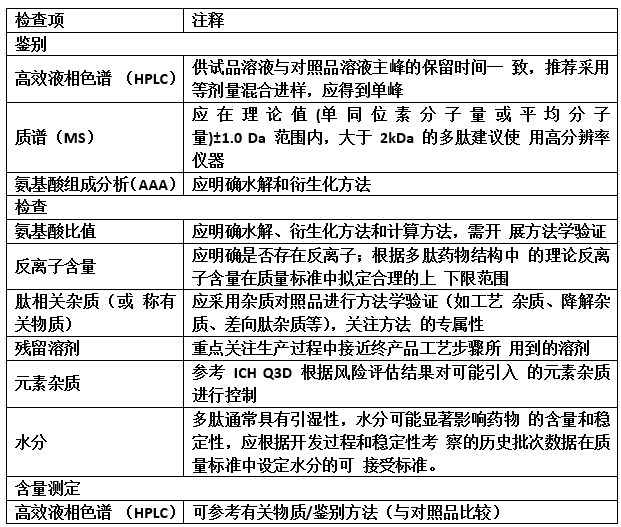
注:注射用原料药建议对微生物限度和细菌内毒素进行研究。
1、鉴别
多肽药物的鉴别方法应具有专属性,通常采用反相高效 液相色谱(RP-HPLC)和质谱法(MS)。此外,还可考虑使 用氨基酸组成分析(AAA)、肽图分析或核磁共振波谱(NMR) 等不同原理的鉴别方法进行正交确认。
2、氨基酸比值 氨基酸分析数据可用于计算多肽药物的氨基酸比值。氨 基酸分析的水解方法可参考相关技术指南进行。通常选择在 水解处理中稳定的氨基酸用于多肽的定量研究,此类氨基酸 主要有门冬氨酸、谷氨酸、丙氨酸、亮氨酸、苯丙氨酸、赖 氨酸和精氨酸。可根据多肽药物的氨基酸序列及不同氨基酸 测定方法调整用于定量研究的氨基酸种类。需明确氨基酸比 值水解方法和计算方法,同时,应进行相应的分析方法验证 工作,证明方法的适用性。
3、杂质 根据化学合成多肽药物的杂质特点,可将其分为肽相关 杂质和非肽杂质。
(1)肽相关杂质
肽相关杂质(或称有关物质)是指与目标分子结构有关 的杂质,是反映多肽化学纯度的重要指标之一。肽相关杂质 可由起始物料引入、生产工艺副反应或长期放置过程中降解 产生。表4列举了肽相关杂质的常见类别及来源,仅作为常见 示例展示,并不能代表全部情况,应结合产品特性进行评估 和分析。
表 4 肽相关杂质的类别及来源
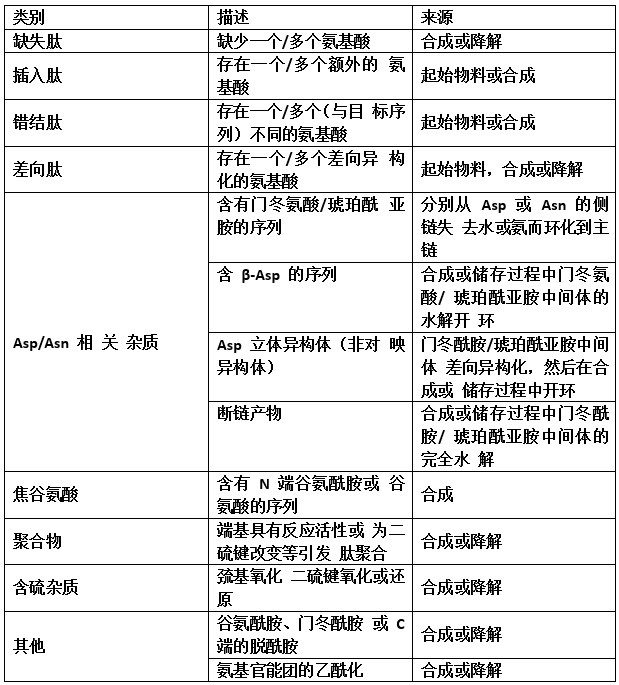
由于部分肽相关杂质的结构、性质与主成分较为相似, 采用单一原理的分析方法可能无法完全分离,推荐采用多种 不同原理的检测方法进行有关物质检查。有关物质检查方法 除常见的高效液相色谱外,还可考虑使用离子交换色谱、毛 细管电泳法等。聚合物检查可采用分子排阻色谱、聚丙烯酰 胺凝胶电泳法等。
差向肽类杂质的色谱行为多与主成分非常接近,鉴定和 分离难度大。可使用氘代试剂水解多肽(例如氯化氘 (DCl)/ 氘代水 (D2O)),采用 Marfey 试剂(FDAA)、邻苯二甲醛 (OPA)或三氟乙酸酐(TFAA)等对水解后的氨基酸进行衍 生,采用相应检测手段测定每个氨基酸手性异构体的含量, 判断出肽链中易发生消旋的氨基酸,进一步制备相应的异构 体杂质对照品进行针对性研究。
有关物质检查方法学验证过程中应重点关注专属性问题,可选择与目标肽结构相近的工艺杂质和降解杂质对照品 进行分离度考察,或采用多肽粗品和强制降解试验样品检验 主峰峰纯度。由于多肽一般为末端吸收,采用 DAD 检测器 无法有效反映峰纯度,可采用液质联用等方法对主峰峰纯度进行研究。结合有关物质项目中确定的特定杂质,采用相应 的杂质对照品进行全面的方法学验证。
本指导原则将肽相关杂质的报告限、鉴定限和质控限分 别制定为 0.1%、0.5%和 1.0%。如发现多肽药物杂质存在安 全性隐患,应适当收紧杂质限度要求。
(2)非肽杂质
非肽杂质是工艺过程中引入的与目标分子结构不相关的 杂质,包括反应试剂和溶剂、金属催化剂、缩合剂等残留物, 此类杂质的研究可参照小分子化学药物相关指导原则进行。
多肽药物在合成过程中,需使用不同类型的缩合剂、保 护基团等,此类物质在反应过程中可能生成含有警示结构的 小分子副产物。应制定合理的控制策略,例如,可采用具有 代表性的化合物或接近终产品工艺步骤生成的化合物进行 针对性的清除效力研究,以证明采用的分离纯化方式可有效 去除相应杂质。
4、含量测定 含量测定是评价多肽质量的重要指标之一,通常在无水、 无反离子的基础上进行研究。一般可采用 HPLC 法,通过与 对照品进行比较,得到目标多肽占总物质的百分比。若经充 分的方法学验证,也可采用其他原理的分析方法。
(二)制剂的质量研究
化学合成多肽制剂的质量研究基本思路和要求可参照小分子化学药物相关指导原则进行,同时提供充分的试验资 料。
多肽药物注射剂的质量标准包括但不限于以下项目:性 状、鉴别、溶液的澄清度与颜色、pH 值、有关物质、装量/装 量差异、可见异物、渗透压摩尔浓度、不溶性微粒、细菌内 毒素、无菌、含量测定等。若处方中添加了抑菌剂、抗氧剂 等,需对抑菌剂含量、抗氧剂含量进行定量检查。
根据多肽药物的稳定性特征和剂型特点,应重点研究制 剂的降解杂质(如脱酰胺、氧化和乙酰化杂质等)、制剂工 艺相关杂质(包括原料药与辅料和/或内包材的反应产物等)。 另外,为降低不良反应发生率,还应对聚合物进行针对性研 究,关注制剂工艺过程、外源因素等的变化对聚合物的影响。
杂质分析方法的选择和相关验证工作可参考原料药部 分进行。
六、稳定性研究
合成多肽药物稳定性研究的基本原则应遵循《化学药物 (原料药和制剂)稳定性研究技术指导原则(修订)》的一般 性要求。结合工艺开发过程中发现的药物不稳定性因素,在 稳定性考察中重点关注。
七、参考文献
[1] ICH Q7: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients. 2000
[2] ICH Q11: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities). 2012
[3] ICH M7 (R1): Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk M7. 2017
[4] 原国家食品药品监督管理局.《合成多肽药物药学研究技 术指导原则》.2007 年 9 月
[5] 原国家食品药品监督管理局.《化学药物制剂研究基本技 术指导原则》.2005 年 3 月
[6] 国家药品监督管理局.《化学药品注射剂仿制药质量和疗 效一致性评价技术要求》.2020 年 5 月
[7] FDA Guidance for Industry. ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin. 2021
[8] USP-NFQuality Attributes of Synthetic Peptide Drug Substances.
[9] USP-NFQuality Attributes of Starting Materials for the Chemical Synthesis of Therapeutic Peptides (PF Online).
[10] Ph.Eur Substances for Pharmaceutical use (monograph 2034).
转载于:https://www.cde.org.cn/main/news/viewInfoCommon/7c105061d4d0f70dfa8e809725a63972